
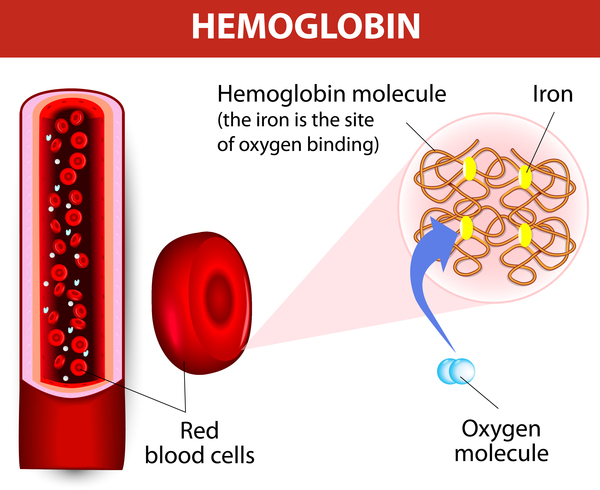
Sickle cell disease is a group of disorders that affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disease have atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape.
Sickle cell disease is a group of inherited red blood cell disorders that affects hemoglobin, the protein that carries oxygen through the body. The condition affects more than 100,000 people in the United States and 20 million people worldwide.
Normally, red blood cells are disc-shaped and flexible to move easily through the blood vessels. If you have sickle cell disease, your red blood cells are crescent or “sickle” shaped. These cells do not bend and move easily and can block blood flow to the rest of your body.
The blocked blood flow through the body can lead to serious problems, including stroke, eye problems, infections, and episodes of pain, called pain crises. Having sickle cell disease also raises your risk for severe illness from COVID-19. Learn the steps you can take to help prevent infection from the Centers for Disease Control and Prevention.
Sickle cell disease is a lifelong illness. A blood and bone marrow transplant is currently the only cure for sickle cell disease, but there are effective treatments that can reduce symptoms and prolong life.
Other Names
- Sickle Cell Trait
- Sickle Cell Crisis
- Exertional Sickling
- Sickling Collapse
- Lumbar Paraspinal Myonecrosis
- Splenic Syndrome
- Splenic Infarction
Pathophysiology
- Sickle cell trait (SCT)
- Inheritance of one gene for sickle hemoglobin (S) and one for normal hemoglobin (A)
- SCT protects against malaria, giving a reproductive advantage to carriers in sub-Saharan Africa
- Each red blood cell (RBC) is about 40% hemoglobin S
- Co-inheritance of alpha thalassemia trait can lower the amount of hemoglobin S (occurs in about 1/3 of African Americans)
What are the types of sickle cell disease?
Hemoglobin is the protein in red blood cells that carries oxygen. It normally has two alpha chains and two beta chains. The four main types of sickle cell anemia are caused by different mutations in these genes.
Hemoglobin SS disease
Hemoglobin SS disease is the most common type of sickle cell disease. It occurs when you inherit copies of the hemoglobin S gene from both parents. This forms hemoglobin known as Hb SS. As the most severe form of SCD, individuals with this form also experience the worst symptoms at a higher rate.
Hemoglobin SC disease
Hemoglobin SC disease is the second most common type of sickle cell disease. It occurs when you inherit the Hb C gene from one parent and the Hb S gene from the other. Individuals with Hb SC have similar symptoms to individuals with Hb SS. However, the anemia is less severe.
Hemoglobin SB+ (beta) thalassemia
Hemoglobin SB+ (beta) thalassemia affects beta globin gene production. The size of the red blood cell is reduced because less beta protein is made. If inherited with the Hb S gene, you will have hemoglobin S beta-thalassemia. Symptoms are not as severe.
Hemoglobin SB 0 (Beta-zero) thalassemia
Sickle beta-zero thalassemia is the fourth type of sickle cell disease. It also involves the beta-globin gene. It has similar symptoms to Hb SS anemia. However, sometimes the symptoms of beta zero thalassemias are more severe. It is associated with a poorer prognosis.
Hemoglobin SD, hemoglobin SE, and hemoglobin SO
These types of sickle cell disease are rare and usually don’t have severe symptoms.
Sickle cell trait
People who only inherit a mutated gene (hemoglobin S) from one parent are said to have sickle cell trait. They may have no symptoms or reduced symptoms.
Diagnosis
History
- Most vital clinical consideration in athletes with SCT because it can be fatal
- Athletes have died in football, basketball, cross-country, track and field, boxing and recreational swimming
- It is the most common cause of death in NCAA Division-1 football (need citation)
- From 2000-2010, all of these deaths occurred during training and conditioning (none during games)
- Part of the reason for NCCAA mandated SCT screening in college athletes
- This phenomenon is also documented among military recruits first noted in 1970
- 4 recruits died, one apparently of coronary heart disease
- The other 3 fit a pattern: collapse during running, hypotension, metabolic acidosis, hyperkalemia, disseminated intravascular coagulation (DIC), oliguria, and “sudden” death 8 to 25 hours after the collapse.
- The US Armed Forces performed a study of all recruits from 1977-to 1981
- The risk of unexplained sudden death in black recruits with SCT was 40 times higher than in all other recruits
- They also found that exercise-related death in black recruits with SCT was 30 times more common than in black recruits without SCT
- Exertional heat illness was often cited as initiating hemostatic dysfunction
- 6 deaths occurred between 1992 and 2001
- One army study looked at 30 exercise collapses of recruits with SCT (need citation)
- The researchers concluded that exertional heat illness was the cause in 22
- However, most had core temperature below 102 or no temperature recorded at all
- Another army study reviewed cases of exertional rhabdomyolysis[6]
- The authors concluded that the main cause of death is “explosive” rhabdomyolysis including hyperkalemia, lactic acidosis, hypocalcemia
- The risk of rhabdomyolysis in recruits with SCT was increased nearly 200-fold
- They cited more often than not sustained maximal exertion rather than exertional heat illness as the cause
Exercise Intensity
- Sickling collapse can occur year-round
- A typical exercise stimulus is a maximal exertion sustained for at least a few minutes
- Can occur from as little sprint distance of only 600 – 1200 m (need citation)
- Can also be due to an abrupt increase in training intensity, training at an unfamiliar altitude or a return to sport with suboptimal physical conditioning
- Of the 10 Division-1 football players who died between 2000 and 2010
- 5 had been performing serial sprints for 5 to 30 minutes
- 4 had been performing fast-tempo, multi-station drills, with little or no rest between stations for 12-60 minutes
- 4 factors are currently thought to contribute: profound hypoxemia, lactic acidosis, hyperosmolarity, and red cell dehydration
- Difficult to distinguish from other more common causes of exertional collapse
- History
- Leg or low back pain, weakness
- Body cramps
- “jello legs” or “my legs won’t go”
- Athletes will slump to the ground or sudden fall or hobble
- Physical exam
- Weakness exceeds the degree of pain
- No evidence of muscle cramping clinically
- Initially lucid and communicative but may have a decline in cognition
- Tachypnea but with clear lungs (from metabolic acidosis)
- Core temperature is typically less than 103
- Controversial
- The army does not currently screen
- All 50 states screen newborns
- NCAA screens all division 1 athletes
Treatment
Stem cell or bone marrow transplants are the only cure for sickle cell disease, but they’re not done very often because of the significant risks involved. Stem cells are special cells produced by bone marrow, a spongy tissue found in the centre of some bones. They can turn into different types of blood cells.
- This a medical emergency and should be treated as such
- Check vital signs including core temperature
- Provide supplemental O2 (if even not hypoxic)
- Cool patient if hyperthermic
- Begin IV hydration
- If not immediate improvement, transfer to hospital via EMS
- Anticipate clinical decline and know where AED is
- Communicate with the emergency department about sickle cell rhabdomyolysis
Medicines
Medicine to prevent the sickling of red blood cells
The U.S. Food and Drug Administration (FDA) approved Voxelotor in 2019 to treat sickle cell disease in adults and children 12 years and older. The oral medicine prevents red blood cells from forming the sickle shape and binding together. This may decrease the destruction of some red blood cells, which in turn lowers the risk for anemia and improves blood flow to your organs.
Possible side effects include headache, diarrhea, abdominal pain, nausea, fatigue, and fever. Rarely, allergic reactions may occur, causing rashes, hives, or mild shortness of breath. Talk to your doctor about other medicines you take.
Medicine to reduce vaso-occlusive and pain crises
In 2019, the FDA approved crizanlizumab-tmca to reduce the number of pain crises experienced by adults and children 16 years and older who have sickle cell disease. The medicine, which is given through an IV in the vein, helps prevent blood cells from sticking to blood vessel walls and causing blood flow blockage, inflammation, and pain crises.
Possible side effects include nausea, joint pain, back pain, and fever.
Hydroxyurea
Hydroxyurea is an oral medicine that has been shown to reduce or prevent several sickle cell disease complications.
- Use in adults: Many studies of adults with hemoglobin SS or hemoglobin Sβ thalassemia showed that hydroxyurea reduced the number of episodes of pain crises and acute chest syndrome. It also improved anemia and decreased the need for transfusions and hospital admissions.
- Use in children: Studies in children with severe hemoglobin SS or Sβ thalassemia showed that hydroxyurea reduced the number of vaso-occlusive crises and hospitalizations. A study of children between the ages of 9 and 18 months with hemoglobin SS or Sβ thalassemia also showed that hydroxyurea reduced the number of pain episodes and dactylitis. There is no information about how safe or effective hydroxyurea is in children under 9 months of age
- Pregnancy: Pregnant people should not use hydroxyurea.
Since hydroxyurea can decrease several complications of sickle cell disease, most experts recommend that children and adults with hemoglobin SS or Sβ0 thalassemia who have frequent painful episodes, recurrent chest crises, or severe anemia take hydroxyurea daily.
Possible side effects include decreased white cell count or platelet count. Rarely, it can worsen anemia. These side effects usually go away quickly if a patient stops taking the medicine. When the patient restarts it, the doctor usually prescribes a lower dose.
It is still unclear whether hydroxyurea can cause problems later in life in people who have sickle cell disease and take the medicine for many years. Studies so far suggest that it does not put people at a higher risk of cancer and does not affect growth in children, but further studies are needed.
Medicine to reduce the risk of infection
In children who have sickle cell disease, taking penicillin two times a day has been shown to reduce the chance of having a severe infection in the bloodstream. Newborns need to take liquid penicillin. Older children can take tablets.
Many doctors will stop prescribing penicillin after a child has reached the age of 5. Some prefer to continue this antibiotic throughout life, particularly if a person has hemoglobin SS or hemoglobin Sβ0 thalassemia since people who have sickle cell disease are still at risk. All people who have had surgical removal of the spleen called a splenectomy, or a past infection with pneumococcus should keep taking penicillin throughout life.
Transfusions
Your doctor may recommend transfusion to treat and prevent certain sickle cell disease complications.
These transfusions may include:
- Acute transfusions treat complications that cause severe anemia. Doctors may also use transfusions when a patient has an acute stroke, in many cases of acute chest crises, and in multi-organ failure. A patient who has sickle cell disease usually receives blood transfusions before surgery, to prevent complications.
- Red blood cell transfusions increase the number of red blood cells and provide normal red blood cells that are more flexible than red blood cells with sickle hemoglobin.
- Regular or ongoing blood transfusions may help lower the chances of another stroke in people who have had an acute stroke.
Doctors also recommend blood transfusions for children who have abnormal transcranial Doppler (TCD) ultrasound results, because transfusions can reduce the chance of having a first stroke. Some doctors use this approach to treat complications that do not improve with hydroxyurea. Doctors may also use transfusions in people who have too many side effects from hydroxyurea. Possible complications include alloimm, which can make it hard to find a matching unit of blood for a future transfusion; infection; and iron overload.
Blood and bone marrow transplant
A blood and bone marrow transplant is currently the only cure for sickle cell disease, but it is not for everyone. Most patients who have sickle cell disease either are too old for a transplant or do not have a relative who is a good enough genetic match to be a donor. A well-matched donor is needed for a patient to have the best chance for a successful transplant.
Most sickle cell disease transplants are currently performed in children who have had complications such as strokes, acute chest crises, and recurring pain crises. These transplants usually use a matched donor. Blood and bone marrow transplants are riskier in adults.
Several medical centers are looking into new ways to help more people who have sickle cell disease get a transplant. These include blood and bone marrow transplant techniques in children and adults who do not have a matched donor in the family or who are older than most recipients.
Blood and bone marrow transplants are successful in about 85% of children when the donor is related and HLA (human leukocyte antigen)-matched. Even with this high success rate, transplants still have risks. Complications can include severe infections, seizures, and other clinical problems. About 5% of people who have received such transplants have died. Sometimes transplanted cells attack the recipient’s organs. This is called graft-versus-host disease. You will get medicine to prevent many of the complications, but they still can happen.
Potential genetic therapy treatments
Researchers at the NHLBI are exploring ways genetic therapies may help develop new treatments or find a cure for sickle cell disease. Genetic therapies aim to treat or cure conditions by adding new DNA or changing existing DNA.
Genetic therapy involves either restoring a faulty or missing gene or adding a new gene that improves the way the cell works. Researchers take blood or bone marrow from a patient and modify their stem cells in a laboratory using genetic therapies.Genetic therapies that modify a person’s own hematopoietic stem cells may provide a cure for people who have sickle cell disease and do not have a well-matched donor. Modified stem cells can be injected into the blood, then the cells travel in the bloodstream to the marrow spaces inside the bones. Once inside the bone marrow, the cells can produce healthy red blood cells that do not sickle.
Prevention
- Allow athletes to set their “own pace”
- The gradual build-up in training rather than an aggressive increase
- No extreme performance tests
- Adjust for heat, altitude
- Control chronic illnesses including asthma
- Maintain adequate hydration
- Disqualify athletes from training if ill
Compartment Syndrome
- Can occur in the lower extremities as seen in non-SCT athletes
- Lumbar Paraspinal Myonecrosis
- Atypical form of acute compartment syndrome in SCT athletes
- Patients complain of acute low back pain with prolonged symptoms for 1-2 weeks
- CK is elevated
- CT and/or MRI is abnormal, show muscle damage, myonecrosis
- Athletes are often able to return to play with modification
Splenic Infarction
- Sometimes referred to as splenic syndrome
- Often associated with exercise at altitude
- Risk thought to begin at 5000 ft, increases at higher altitudes
- The famous case of Ryan Clark, former safety for the Pittsburg Steelers
- Played at elevation (Denver, CO) in 2005 and was diagnosed with “splenic contusion”
- Played again in 2007 and suffered a splenic infarction, including an abscess requiring splenectomy and cholecystectomy
Hematuria
- Overall, rare and occurs in fewer than 5% of patients with SCT (need citation)
- Case reports of hematuria attributed to SCT
- Somewhat a diagnosis of exclusion after normal IV pyelogram and cystoscopy
- Results from sickling deep within the renal medulla
- Occasionally associated with papillary necrosis
- 80% from left kidney
- Treatment is generally conservative and includes
- Relative rest
- Hydration
- Iron supplementation as needed
- For progressive or persistent hematuria, can consider
- Vasopressin
- Aminocaproic acid
- The efficacy of these agents is unknown
- Hyposthenuria is the inability to concentrate urine
- Occurs in SCT due to altered blood flow to the renal medulla
- Clinical Significance is unknown
Venous Thromboembolism (VTE)
- SCT increases the relative risk of pulmonary embolism by 1.5 – 2.0
- For reasons that are less clear, the risk of DVT may not be as great
References







{kind=link}